Publications: *: Corresponding author
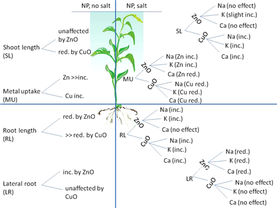
11. Jacob Stewart, Trevor Hansen, Joan E McLean, Paul McManus, Sid Das, David W Britt, Anne J Anderson, Christian O Dimkpa; "Salts affect the interaction of ZnO or CuO nanoparticles with wheat" Environmental Toxicology and Chemistry, 2015, 34, 2116-2125.
Abstract. Exposure to nanoparticles (NPs) that release metals with potential phytotoxicity could pose problems in agriculture. The authors of the present study used growth in a model growth matrix, sand, to examine the influence of 5 mmol/kg of Na, K, or Ca (added as Cl salts) and root exudates on transformation and changes to the bioactivity of copper(II) oxide (CuO) and zinc oxide (ZnO) NPs on wheat.
Abstract. Exposure to nanoparticles (NPs) that release metals with potential phytotoxicity could pose problems in agriculture. The authors of the present study used growth in a model growth matrix, sand, to examine the influence of 5 mmol/kg of Na, K, or Ca (added as Cl salts) and root exudates on transformation and changes to the bioactivity of copper(II) oxide (CuO) and zinc oxide (ZnO) NPs on wheat.
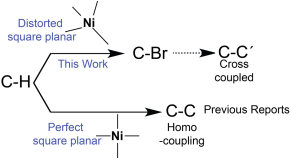
10. Moumita Bhattacharya, David Cluff and Sid Das*; "Ni(II) Catalyzed Bromination of Aryl C-H Bonds" Inorganica Chimica Acta, 2014, 423, 238-241.
Abstract. Bromination of unactivated aromatic C–H bonds without directing and/or chelating groups was achieved by employing an air stable N-heterocyclic Ni(II) complex. PhI(OAC)2 and N-bromosuccinimide have been used as the oxidizing agent and the bromine source, respectively. Yields for bromination are as high as >99%, especially in presence of electron-withdrawing groups like –NO2 and –CF3. This is a rare report on C–H bond activation with Ni(II) where, instead of homo C–C coupling, reductive elimination to form C-halogen could be achieved.
Abstract. Bromination of unactivated aromatic C–H bonds without directing and/or chelating groups was achieved by employing an air stable N-heterocyclic Ni(II) complex. PhI(OAC)2 and N-bromosuccinimide have been used as the oxidizing agent and the bromine source, respectively. Yields for bromination are as high as >99%, especially in presence of electron-withdrawing groups like –NO2 and –CF3. This is a rare report on C–H bond activation with Ni(II) where, instead of homo C–C coupling, reductive elimination to form C-halogen could be achieved.
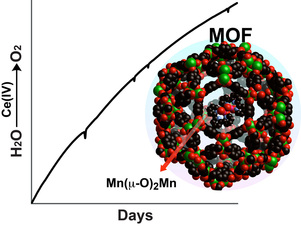
9. Rebecca E. Hansen and Sid Das*; "Biomimetic Di-Manganese Catalyst Cage-Isolated in a MOF: Robust Catalyst for Water Oxidation with Ce(IV), a non-O-donating oxidant" Energy and Environmental Science, 2014, 7, 317-322.
Abstract: A biomimetic di-Mn catalyst catalyzes water oxidation with (NH4)2Ce(NO3)6 for >7 days with initial TOF of 40 per hr. Isolation of one molecule of catalyst in each pore of a Cr-based MOF prevents decomposition of the catalyst and allows sustained water oxidation even at pH 1.00.
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2013/EE/C3EE43040E#!divAbstract
Abstract: A biomimetic di-Mn catalyst catalyzes water oxidation with (NH4)2Ce(NO3)6 for >7 days with initial TOF of 40 per hr. Isolation of one molecule of catalyst in each pore of a Cr-based MOF prevents decomposition of the catalyst and allows sustained water oxidation even at pH 1.00.
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2013/EE/C3EE43040E#!divAbstract
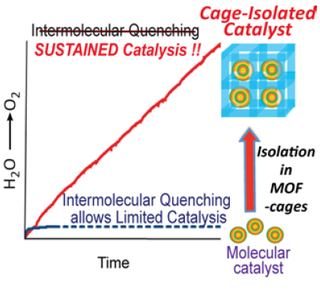
8. Binod Nepal and Sid Das*; “Sustained Water Oxidation by a Catalyst Cage-Isolated in a Metal–Organic Framework” Angew. Chem. Int. Ed., 2013, 52, 7224-7227.
(See highlight in Chemical & Engineering News:
http://cen.acs.org/articles/91/i26/Stable-Water-Oxidation-Inside-Cage.html)
Abstract: Pores of a MOF were used to cage-isolate a highly reactive water-oxidation catalyst (Mn(μ-O)2Mn). The resulting construct showed highly sustained water oxidation, with minimized inter-molecular degradative side-reactions. Our system provides a simple path to extend the lifetime of reactive catalysts without compromising their molecular definition and reactivity.
Web-link: http://onlinelibrary.wiley.com/doi/10.1002/anie.201301327/abstract
(See highlight in Chemical & Engineering News:
http://cen.acs.org/articles/91/i26/Stable-Water-Oxidation-Inside-Cage.html)
Abstract: Pores of a MOF were used to cage-isolate a highly reactive water-oxidation catalyst (Mn(μ-O)2Mn). The resulting construct showed highly sustained water oxidation, with minimized inter-molecular degradative side-reactions. Our system provides a simple path to extend the lifetime of reactive catalysts without compromising their molecular definition and reactivity.
Web-link: http://onlinelibrary.wiley.com/doi/10.1002/anie.201301327/abstract
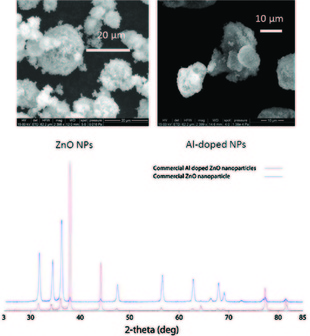
7. Tommy Fang, Jean-Lic Watson, Jordan Goodman, Christian Dimpka, Nicole Martineau, Sid Das, Joan McLean, David Britt, Anne Anderson; “Does doping with aluminum alter the effects of ZnO nanoparticles on themetabolism of soil pseudomonads?” Microbiological Research, 2013, 168, 91-98.
Abstract. Doping of ZnO nanoparticles (NPs) is being used to increase their commercialization in the optical and semiconductor fields. This paper addresses whether doping with Al alters how NPs at nonlethal levels modify the metabolism of soil-borne pseudomonads. Differences in XRD, absorbance and fluorescent spectra were observed between commercial ZnO and Al-doped ZnO NPs. Both particles aggregated in the bacterial growth medium and formed colloids of different surface charges. They had similar effects on bacterial metabolism: rapid, dose-dependent loss in light output indicative of temporary toxicity in a biosensor constructed in Pseudomonas putida KT2440; increased production of a fluorescent pyoverdine-type siderophore, and decreased levels of indole acetic acid and phenazines in P. chlororaphis O6. Solubilization of Zn and Al from the NPs contributed to these responses to different extents. These findings indicate that Al-doping of the ZnO NPs did not reduce the ability of the NPs to alter bacterial metabolism.
Web-link: http://www.sciencedirect.com/science/article/pii/S0944501312001012
Abstract. Doping of ZnO nanoparticles (NPs) is being used to increase their commercialization in the optical and semiconductor fields. This paper addresses whether doping with Al alters how NPs at nonlethal levels modify the metabolism of soil-borne pseudomonads. Differences in XRD, absorbance and fluorescent spectra were observed between commercial ZnO and Al-doped ZnO NPs. Both particles aggregated in the bacterial growth medium and formed colloids of different surface charges. They had similar effects on bacterial metabolism: rapid, dose-dependent loss in light output indicative of temporary toxicity in a biosensor constructed in Pseudomonas putida KT2440; increased production of a fluorescent pyoverdine-type siderophore, and decreased levels of indole acetic acid and phenazines in P. chlororaphis O6. Solubilization of Zn and Al from the NPs contributed to these responses to different extents. These findings indicate that Al-doping of the ZnO NPs did not reduce the ability of the NPs to alter bacterial metabolism.
Web-link: http://www.sciencedirect.com/science/article/pii/S0944501312001012
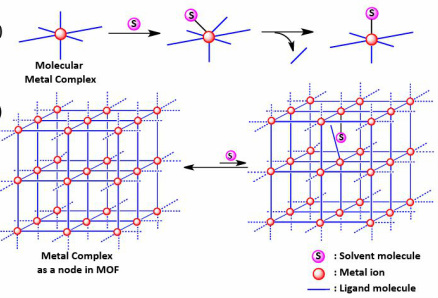
6. Sanjit Das, Daniel Johnston, Sid Das*; “Structural Bolstering of Metal Sites as Nodes in Metal Organic Frameworks”. CrystEngComm, 2012, 14, 6136-6139.
(among “Top 10 Highest Accessed Articles” in August, 2012)
Abstract: We show that a metal site attains orders of magnitude higher resistance (compared to its molecular form) towards external ligand-induced disruption of its coordination environment upon incorporation as the building block of a crystalline metal–organic framework (MOF).
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2012/CE/C2CE25555C
(among “Top 10 Highest Accessed Articles” in August, 2012)
Abstract: We show that a metal site attains orders of magnitude higher resistance (compared to its molecular form) towards external ligand-induced disruption of its coordination environment upon incorporation as the building block of a crystalline metal–organic framework (MOF).
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2012/CE/C2CE25555C
Publications from Graduate work (Ph.D.):
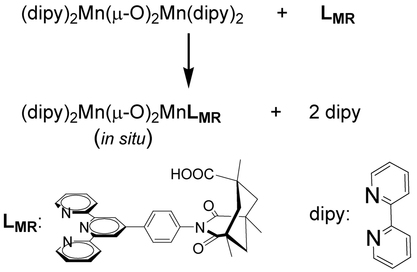
5. Sid Das, Gary W. Brudvig*, Robert H. Crabtree*; “In situ dimanganese catalyst for fast screening of molecular recognition catalysts for regioselective oxygenation of an sp3 C-H bond”, Inorg. Chim. Acta 2009, 362, 1229.
Abstract: We report a rapid method for assembling our di-μ-oxo dimanganese catalyst, verified by ESI-MS and EPR, assessing its water oxidation activity by a Clark electrode O2-assay study and its regioselective C–H activation activity by product analysis in catalytic runs.
Web-link: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2898201/
Abstract: We report a rapid method for assembling our di-μ-oxo dimanganese catalyst, verified by ESI-MS and EPR, assessing its water oxidation activity by a Clark electrode O2-assay study and its regioselective C–H activation activity by product analysis in catalytic runs.
Web-link: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2898201/
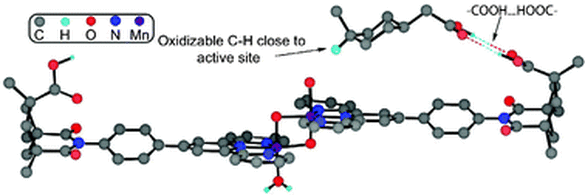
4. Sid Das, Gary W. Brudvig*, Robert H. Crabtree*; “Molecular recognition in homogeneous transition metal catalysis: a biomimetic strategy for high selectivity”, Chem. Comm. 2008, 413-424.
Abstract: Traditional methods for selectivity control in homogeneous transition metal catalysis either employ steric effects in a binding pocket or chelate control. In a supramolecular strategy, encapsulation of the substrate can provide useful shape and size selectivity. A fully developed molecular recognition strategy involving hydrogen bonding or solvophobic forces has given almost completely regioselective functionalization of remote, unactivated C–H bonds.
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2008/CC/b710355g
Abstract: Traditional methods for selectivity control in homogeneous transition metal catalysis either employ steric effects in a binding pocket or chelate control. In a supramolecular strategy, encapsulation of the substrate can provide useful shape and size selectivity. A fully developed molecular recognition strategy involving hydrogen bonding or solvophobic forces has given almost completely regioselective functionalization of remote, unactivated C–H bonds.
Web-link: http://pubs.rsc.org/en/Content/ArticleLanding/2008/CC/b710355g
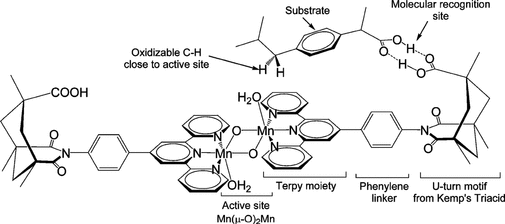
3. Sid Das, Gary W. Brudvig*, Robert H. Crabtree*; “High turnover remote catalytic oxygenation of alkyl groups: the role of self-Inhibition for molecular recognition selectivity”, J. Am. Chem. Soc. 2008, 130, 1628-1637.
Abstract: H-bonding mediated molecular recognition between substrate and ligand −COOH groups orients the substrate so that remote, catalyzed oxygenation of an alkyl C−H bond by a Mn-oxo active site can occur with very high (>98%) regio- and stereoselectivity. This paper identifies steric exclusionexclusion of non H-bonded substrate molecules from the active siteas one requirement for high selectivity, along with the entropic advantage of intramolecularity. If unbound substrate molecules were able to reach the active site, they would react unselectively, degrading the observed selectivity. Both of the faces of the catalyst are blocked by two ligand molecules each with a −COOH group. The acid p-tBuC6H4COOH binds to the ligand −COOH recognition site but is not oxidized and merely blocks approach of the substrate therefore acting as an effective inhibitor for ibuprofen oxidation in both free acid and ibuprofen ester form. Dixon plots show that inhibition is competitive for the free acid ibuprofen substrate, no doubt because this substrate can compete with the inhibitor for binding to the recognition site. In contrast, inhibition is uncompetitive for the ibuprofen-ester substrate, consistent with this ester substrate no longer being able to bind to the recognition site. Inhibition can be reversed with MeCOOH, an acid that can competitively bind to the recognition site but, being sterically small, no longer blocks access to the active site.
Web-link: http://pubs.acs.org/doi/abs/10.1021/ja076039m
Abstract: H-bonding mediated molecular recognition between substrate and ligand −COOH groups orients the substrate so that remote, catalyzed oxygenation of an alkyl C−H bond by a Mn-oxo active site can occur with very high (>98%) regio- and stereoselectivity. This paper identifies steric exclusionexclusion of non H-bonded substrate molecules from the active siteas one requirement for high selectivity, along with the entropic advantage of intramolecularity. If unbound substrate molecules were able to reach the active site, they would react unselectively, degrading the observed selectivity. Both of the faces of the catalyst are blocked by two ligand molecules each with a −COOH group. The acid p-tBuC6H4COOH binds to the ligand −COOH recognition site but is not oxidized and merely blocks approach of the substrate therefore acting as an effective inhibitor for ibuprofen oxidation in both free acid and ibuprofen ester form. Dixon plots show that inhibition is competitive for the free acid ibuprofen substrate, no doubt because this substrate can compete with the inhibitor for binding to the recognition site. In contrast, inhibition is uncompetitive for the ibuprofen-ester substrate, consistent with this ester substrate no longer being able to bind to the recognition site. Inhibition can be reversed with MeCOOH, an acid that can competitively bind to the recognition site but, being sterically small, no longer blocks access to the active site.
Web-link: http://pubs.acs.org/doi/abs/10.1021/ja076039m
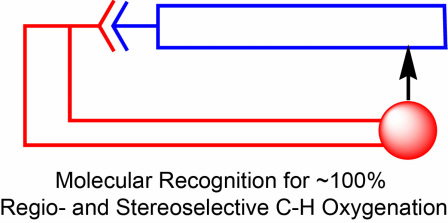
2. Sid Das, Chris D. Incarvito, Robert H. Crabtree*, Gary W. Brudvig*; “Molecular Recognition in the Selective Oxygenation of Saturated C-H Bonds by a Dimanganese Catalyst”, Science 2006, 312, 1941-1943.
(See highlight in Science 2006, 312, 1885-1886 and in Chemical & Engineering News 2006, 84, issue 27, p. 11)
A new catalytic design that comprises a manganese oxo active site with a molecular recognition group to provide highly regioselective C-H oxidation of organic substrates.
Abstract: Although enzymes often incorporate molecular recognition elements to orient substrates selectively, such strategies are rarely achieved by synthetic catalysts. Here we combine molecular recognition via hydrogen bonding with C-H activation to obtain high-turnover catalytic regioselective functionalization of sp3 C-H bonds remote from the –COOH recognition group. The catalyst contains a Mn(μ-O)2Mn reactive center and a ligand based on Kemp’s triacid that directs a –COOH group to anchor the carboxylic acid group of the substrate and thus modify the usual selectivity for oxidation. Control experiments support the role of hydrogen bonding in orienting the substrate to achieve high selectivity.
Web-link: https://www.sciencemag.org/content/312/5782/1941.abstract
(See highlight in Science 2006, 312, 1885-1886 and in Chemical & Engineering News 2006, 84, issue 27, p. 11)
A new catalytic design that comprises a manganese oxo active site with a molecular recognition group to provide highly regioselective C-H oxidation of organic substrates.
Abstract: Although enzymes often incorporate molecular recognition elements to orient substrates selectively, such strategies are rarely achieved by synthetic catalysts. Here we combine molecular recognition via hydrogen bonding with C-H activation to obtain high-turnover catalytic regioselective functionalization of sp3 C-H bonds remote from the –COOH recognition group. The catalyst contains a Mn(μ-O)2Mn reactive center and a ligand based on Kemp’s triacid that directs a –COOH group to anchor the carboxylic acid group of the substrate and thus modify the usual selectivity for oxidation. Control experiments support the role of hydrogen bonding in orienting the substrate to achieve high selectivity.
Web-link: https://www.sciencemag.org/content/312/5782/1941.abstract
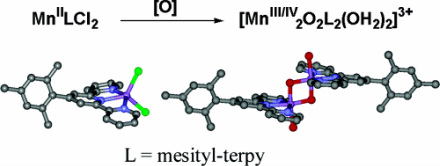
1. Hongyu Chen, Ranitendranath Tagore, Sid Das, Chris D. Incarvito, Jack W. Faller, Robert H. Crabtree*, Gary W. Brudvig*; “General Synthesis of Di-μ-oxo Dimanganese Complexes as Functional Models for the Oxygen Evolving Complex of Photosystem II”, Inorg. Chem. 2005, 44, 7661-7670.
Abstract: A series of complexes with the formula [MnIII/IV2(μ-O)2(L)2(X)2]3+ have been prepared in situ from MnIILCl2 precursors by a general preparative method (L = terpy, Cl-terpy, Br-terpy, Ph-terpy, tolyl-terpy, mesityl-terpy, t Bu3-terpy, EtO-terpy, py-phen, dpya, Me2N-terpy, or HO-terpy, and X = a labile ligand such as water, chloride, or sulfate). The parent complex, where L = terpy and X = water, is a functional model for the oxygen-evolving complex of photosystem II (Limburg, et al. J. Am. Chem. Soc. 2001, 123, 423−430). Crystals of MnII(dpya)Cl2, MnII(Ph-terpy)Cl2, MnII(mesityl-terpy)Cl2, and an organic-soluble di-μ-oxo di-aqua dimanganese complex, [MnIII/IV2(μ-O)2(mesityl-terpy)2(OH2)2](NO3)3, were obtained and characterized by X-ray crystallography. Solutions of the in situ-formed di-μ-oxo dimanganese complexes were characterized by electrospray mass spectrometry, EPR spectroscopy, and UV−visible spectroscopy, and the rates of catalytic oxygen-evolving activity were assayed. The use of MnIILCl2 precursors leads to higher product purity of the Mn dimers while achieving the 1:1 ligand to Mn stoichiometry appropriate for catalytic activity assay. These methods can be used to screen the catalytic activity of other di-μ-oxo dimanganese complexes generated by using a ligand library.
Web-link: http://pubs.acs.org/doi/abs/10.1021/ic0509940
Abstract: A series of complexes with the formula [MnIII/IV2(μ-O)2(L)2(X)2]3+ have been prepared in situ from MnIILCl2 precursors by a general preparative method (L = terpy, Cl-terpy, Br-terpy, Ph-terpy, tolyl-terpy, mesityl-terpy, t Bu3-terpy, EtO-terpy, py-phen, dpya, Me2N-terpy, or HO-terpy, and X = a labile ligand such as water, chloride, or sulfate). The parent complex, where L = terpy and X = water, is a functional model for the oxygen-evolving complex of photosystem II (Limburg, et al. J. Am. Chem. Soc. 2001, 123, 423−430). Crystals of MnII(dpya)Cl2, MnII(Ph-terpy)Cl2, MnII(mesityl-terpy)Cl2, and an organic-soluble di-μ-oxo di-aqua dimanganese complex, [MnIII/IV2(μ-O)2(mesityl-terpy)2(OH2)2](NO3)3, were obtained and characterized by X-ray crystallography. Solutions of the in situ-formed di-μ-oxo dimanganese complexes were characterized by electrospray mass spectrometry, EPR spectroscopy, and UV−visible spectroscopy, and the rates of catalytic oxygen-evolving activity were assayed. The use of MnIILCl2 precursors leads to higher product purity of the Mn dimers while achieving the 1:1 ligand to Mn stoichiometry appropriate for catalytic activity assay. These methods can be used to screen the catalytic activity of other di-μ-oxo dimanganese complexes generated by using a ligand library.
Web-link: http://pubs.acs.org/doi/abs/10.1021/ic0509940